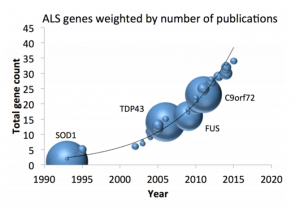
 Approximately 15% of all ALS patients have a family history of ALS or frontotemporal dementia. So far, defects in multiple genes have been identified to cause familial ALS, including the SOD1, C9orf72,TDP43 and FUS genes. This month, New England Journal of Medicine published two studies on genomic therapies for ALS patients carrying SOD1 mutations.
Approximately 15% of all ALS patients have a family history of ALS or frontotemporal dementia. So far, defects in multiple genes have been identified to cause familial ALS, including the SOD1, C9orf72,TDP43 and FUS genes. This month, New England Journal of Medicine published two studies on genomic therapies for ALS patients carrying SOD1 mutations.
The first study describes the results of an early phase clinical trial (phase 1) to evaluate the drug ‘tofersen’. This drug is administered via intrathecal injections (injections in the lower back to reach the cerebrospinal fluid, the fluid in and around the brain and spinal cord). While the aim of this study was to evaluate the safety of the treatment, there is promising preliminary data that suggests the drug may be effective. The researchers found that the highest dose of tofersen decreased SOD1 protein levels in the cerebrospinal fluid (a fluid in and around the brain and spinal cord). In addition, there is indirect evidence that the drug diminishes the loss of brain cells and functional decline in ALS-patients carrying SOD1 mutations, especially those with a fast-progressing disease course. Currently, a phase 3 clinical trial is already ongoing to confirm these findings over a longer follow-up period.
In the second study, two patients received a single dose of a different gene-silencing therapy. Although SOD1 protein levels in the cerebrospinal fluid remained unchanged, a post-mortem analysis showed that SOD1 protein levels were decreased in the spinal cord of one of the patients. The first patient to be treated with this novel therapy developed meningoradiculitis (a form of meningitis which also involves nerve roots leading to intense nerve pain radiating from the spine), which was prevented in the second patient using immunosuppressive drugs. While no conclusion can be drawn regarding the safety and efficacy of this treatment based on this study, it does suggest that this method of gene silencing is worth exploring further.
 These two landmark studies demonstrate that genetic discoveries can directly lead to the development of genomic therapies for ALS patients. This again reinforces the relevance of Project MinE to patients with ALS. Project MinE therefore aims to continue improving our understanding regarding the genetic risk factors for underlying ALS. To overall goal of the project is to fully map the complete DNA profiles of at least 15,000 people with ALS and compare these to at least 7,500 healthy controls.
These two landmark studies demonstrate that genetic discoveries can directly lead to the development of genomic therapies for ALS patients. This again reinforces the relevance of Project MinE to patients with ALS. Project MinE therefore aims to continue improving our understanding regarding the genetic risk factors for underlying ALS. To overall goal of the project is to fully map the complete DNA profiles of at least 15,000 people with ALS and compare these to at least 7,500 healthy controls.
At present, over 10,000 genomes have been sequenced in Project MinE and this has already led to the discovery of multiple new ALS genes, such as NEK1, C21orf2, NIPA1, ATXN1 and KIF5a. By continuing to identify novel genetic risk factors for ALS, we aim to drive the development of targeted and effective therapies.